- Credits
- Section Writer: Dr. Om J Lakhani
- Section Editor: Dr. Om J Lakhani
Support us:
- Support us by purchasing our book - Click here for more details: Volume 1- THE BEST OF NOTES IN ENDOCRINOLOGY BOOK SERIES
- Support you by Becoming a YouTube member (Click here)
- Q. What are the recommendations for genetic screening for pheochromocytoma?
- Endocrine society guidelines – it should be offered to all patients as a part of shared decision making
- Q. How common are pheochromocytomas hereditary?
- 30-40% are hereditary in adults
- More than 80% are hereditary in children
- Pearl
- Catecholamine-secreting pheochromocytoma are more likely to be bilateral
- Q. Enlist the various genes associated with pheochromocytoma.
- RET
- NF1
- VHL
- SDHAF2
- SDHA
- SDHB
- SDHC
- SDHD
- TMEM127
- MAX
- FH
- EPAS1
- MDH2
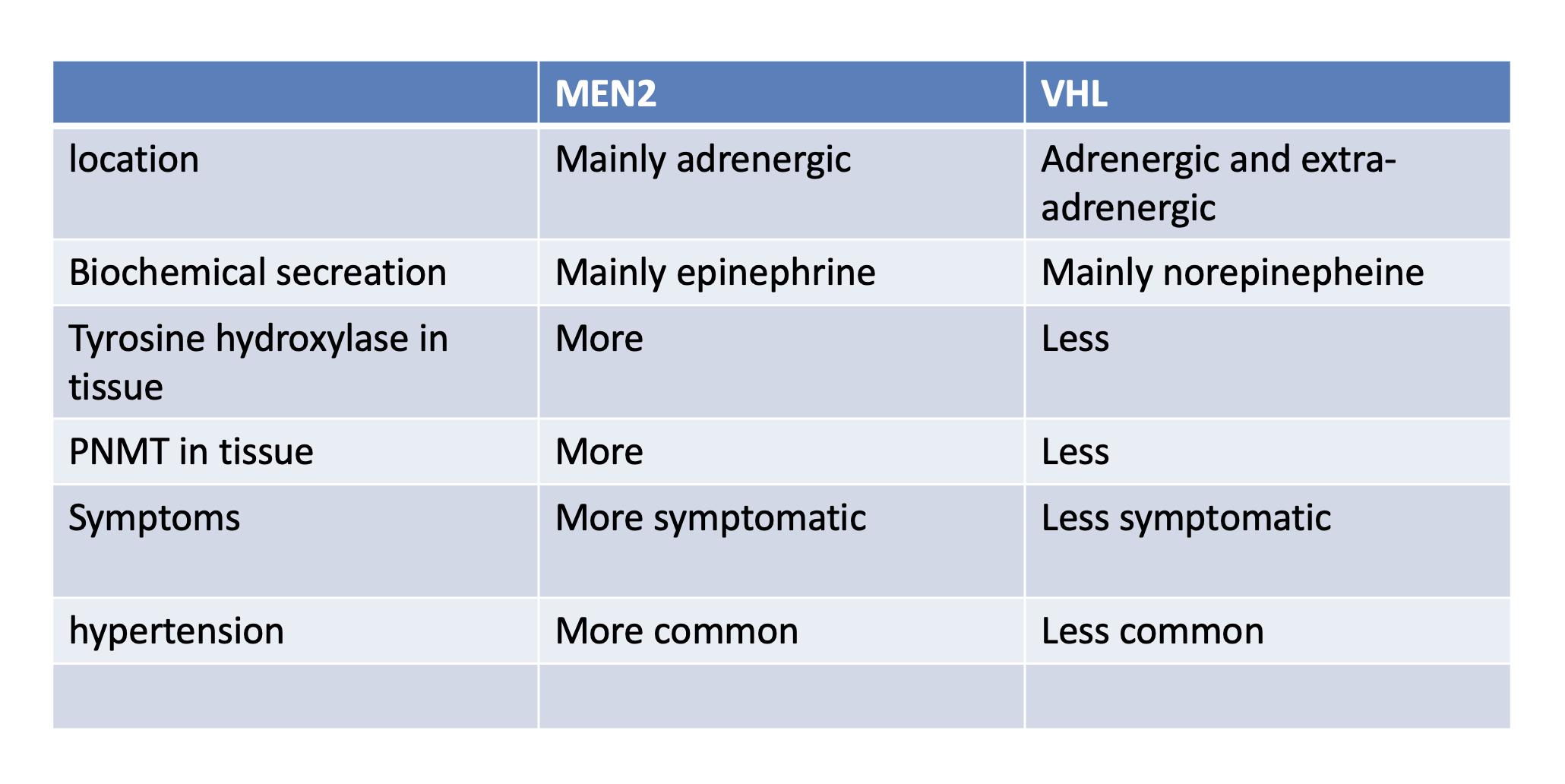
- Q. How common is pheochromocytoma in each of these syndromes – MEN2, VHL, and Neurofibromatosis?
- VHL- 10-20%
- MEN 2- 50%
- NF 1 – 0.1-6%
- Von hippel lindau
- Pearl- PHEOCHROMOCYTOMA occurs under the age of 30 in these people
- Q. What are the clinical features of VHL?
- Hemangioblastoma- Cerebellum, Spinal cord, or brainstem
- Pheochromocytoma
- Paraganglioma
- Retinal angioma
- Clear cell carcinoma
- Pancreatic NET
- Endolymphatic sac tumors of the middle ear
- Serous cystadenoma of the pancreas
- Papillary cystadenoma of epididymis and broad ligament
- Q. What does the VHL gene do?
- It is a tumor suppressor gene
- Q. Where is it located?
- 3p25-26
- Q. What are the subtypes of VHL?

- Type II VHL- higher risk of Pheochromocytoma
- Q. Which subgroup of VHL mutation has more risk of Pheochromocytoma?
- Missense mutation higher likelihood of pheochromocytoma
- Q. When should screening for pheochromocytoma start in VHL patients?
- At age 5 years
- Q. What is the characteristic of Pheochromocytoma in VHL?
- Bilateral
- Adrenal
- Norepinephrine secreting
- (most adrenal pheochromocytoma secrete epinephrine, this one is unusual)
- Q. What about extra-adrenal, do VHL have paraganglioma also?
- Yes
- They may have paraganglioma also
- MEN 2
- Q. How common is pheochromocytoma in MEN2?
- 50% of cases
- Q. Are they adrenal or extradrenal?
- Mainly adrenal
- Q. Which are the high-risk mutations in MEN 2?
- Codon
- 634
- 918
- 883
- Codon
- Q. When should surveillance for pheochromocytoma begin in high-risk mutations?
- New guidelines- At age 11
- Q. What about moderate risk mutations?
- Age 16
- Q. What are the features of MEN 2A?
- MTC- 100%
- Pheochromocytoma – 50%
- Hyperparathyroidism – 20%
- Cutaneous lichen amyloidosis
- Hirscpung disease- having Janus mutation
- Q. Which is the commonest mutation associated with MEN2A?
- In codon 634- in 85% of cases
- Q. What are the clinical features of MEN 2B?
- MTC- in all
- Pheochromocytoma – 50%
- Mucocutaneous neuromas- tongue, lips, and eyelid
- Joint laxity
- Skeletal deformities- lordosis or kyphoscoliosis
- Intestinal ganglioneuroma
- Q. Give the difference in pheochromocytoma between VHL and MEN2.
- **Neurofibromatosis type 1 **
- Q. What are the diagnostic criteria for NF1?
- Any two of below
- 6 or more Café-au-luit spots
- 2 or more Lisch nodule
- 2 or more cutaneous neurofibroma
- Sphenoid dysplasia
- Optic glioma
- Axillary Freckling
- Relative having NF1
- Any two of below
- Q. What is the prevalence of Pheochromocytoma in NF1?
- 0.5-6%
- Q. Which NF1 patients must be screened for PCC?
- Only those with hypertension
- Q. What type of PCC tumors are common in NF1?
- Bilateral and mainly adrenal
- Q. What kind of biochemical release is common with NF1 and RET?
- Epinephrine predominance
- SDH mutation
- Q. Describe the SDH mutation based on the PGL numbering.
- PGL1- SDHD
- PGL2- SDHAF2
- PGL3- SDHC
- PGL4- SDHB
- PGL5- SDHA
- Q. Which SDH mutation are Autosomal dominant ?
- SDHB or SDHC
- Q. Which are SDH mutations paternally inherited?
- SDHD or SDHAF2 (maternal imprinting)
- Q. Which SDH has a higher risk of metastasis?
- SDHB – B for bad
- About 20-30% risk of Mets
- Other is less than 5%
- Q. Summarize the clinical manifestations of various SDH mutations.
- EA PGL- extraadrenal paraganglioma
- PCC- pheochromocytoma
- HNPGL- head and neck paraganglioma
- Q. How common is renal cell carcinoma in patients with SDHB mutation?
- Very common
- 30-40%
- Q. Summarize the lesions seen with SDHB mutation.
- Metastatic Paraganglioma
- Head and neck paraganglioma
- Pheochromocytoma
- Renal cell carcinoma (common)
- Pituitary adenoma
- GIST
- Q. Which SDH produces head and neck PGL?
- SDHAF2, SDHB, SDHC and SDHD (all except SHDA)
- Q. What are the features of head and neck PGL?
- They are often asymptomatic
- Sometimes secrete a parasympathetic neurotransmitter
- Q. Which is most commonly associated with Pituitary adenoma?
- SDHD
- Q. Enlist the other tumors associated with SDH.
- Renal cell carcinoma (clear cell carcinoma) - SDHB
- GIST – SDHA
- Pituitary adenoma – usually SDHD
- Q. Which is the commonest location for the paragangliomas?
- Abdominal
- Q. Which biochemistry alone is not enough in the above case?
- Many PPGLs may be non-secreting
- To pick up other tumors associated with SDHB like renal cell carcinoma
- Q. If metastasis is considered in this case- is PET better or MIBG better?
- PET is better
- Q. How frequently should you do biochemistry and imaging?
- No guidelines
- Annual biochemistry
- MRI/CT- every 2 years
- Q. What do you do for asymptomatic patients picked up with SDHB mutation?
- Annual biochemistry + 2 Year imaging
- Youngest age may be age 5
- However, we do not have population-based data
- Q. How long to follow up?
- Lifelong
- Pearl
- Head and neck paraganglioma are usually non-secretory
- If there is a secretory pattern in a patient with a history of H and N PPGL- look for tumors elsewhere
- Pearl
- Patients with pheochromocytoma ANY procedure → preparation similar to surgery is required
- **Genetic screening **
- Q. Which patients are more likely to have a genetic cause of pheochromocytoma?
- Age of onset <45 years
- Bilateral
- Extra-adrenal
- Family history
- Syndromic features present
- **Others **
- Q. Which are cluster 1 and cluster 2 genes associated with pheochromocytoma?
- Cluster 1 – genes dealing with hypoxia
- Cluster 2- genes dealing with kinase signaling
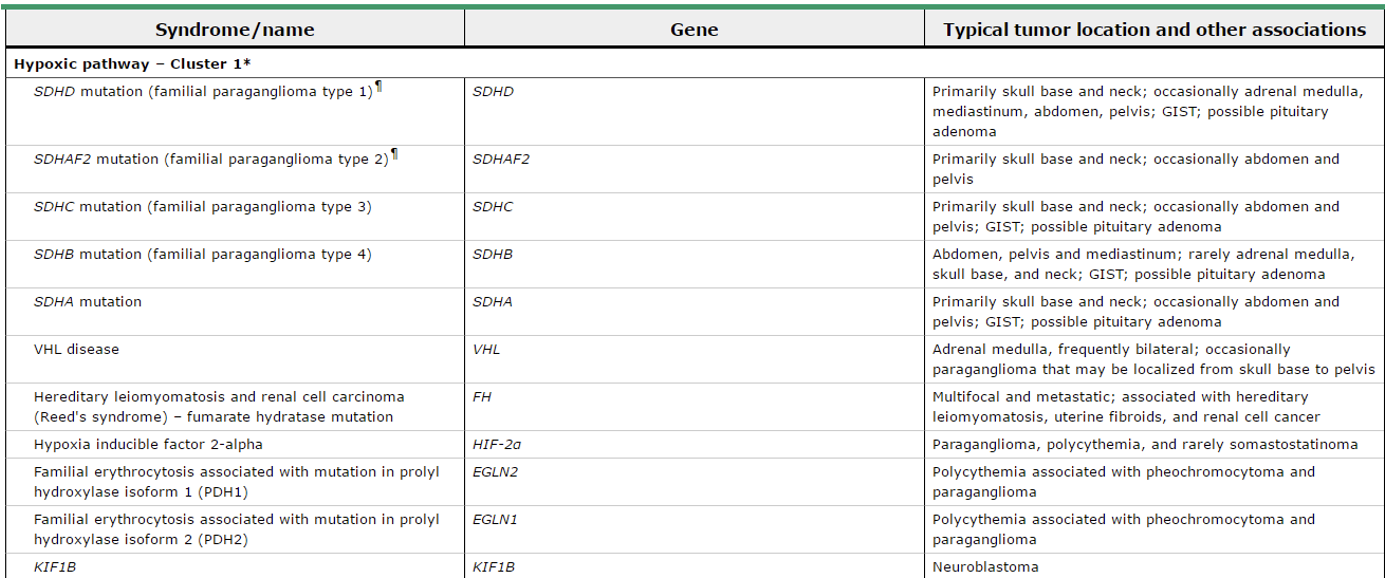

- Q. What is the difference between cluster 1 and cluster 2 genes?
- MUTATION TESTING ALGORITHM
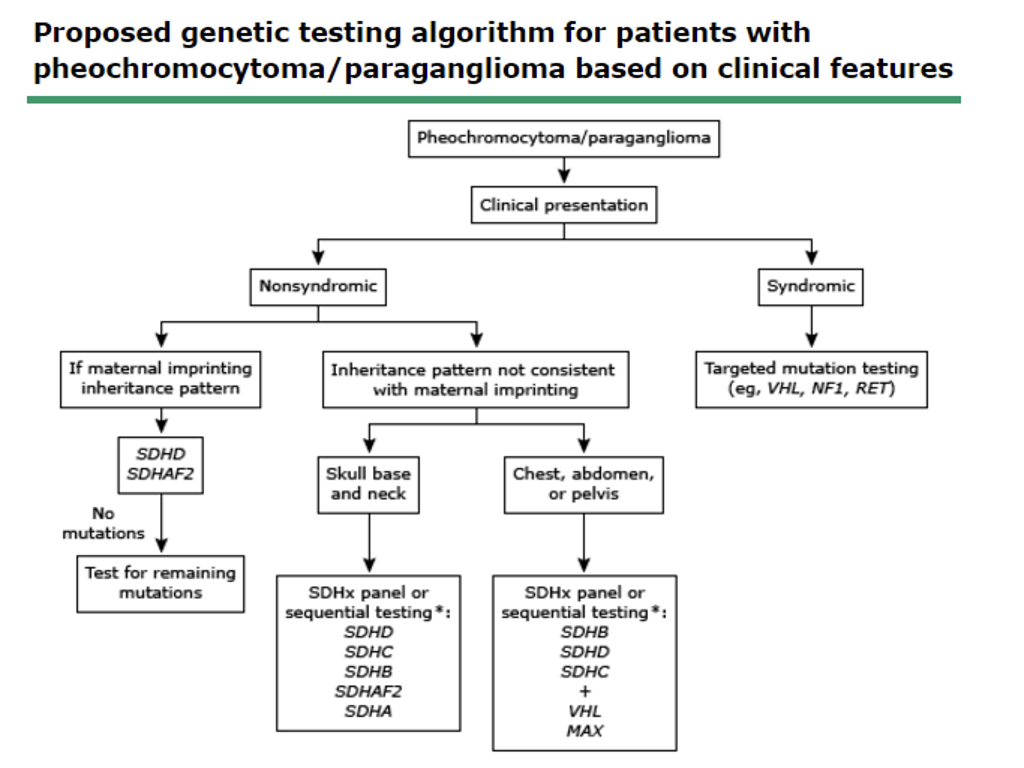
- Q. Which mutation to test first in case of malignancy?
- SDHB
- Q. Which mutation to test in the case of head and neck PPGL?
- SDHD
- SDHC
- SDHAF2
- Q. Which mutation to test in the case of dopaminergic?
- SDHB
- Q. Which mutation to test in case of extra-adrenal Norepinephrine?
- SDHB
- Q. What about adrenal but norepinephrine?
- VHL
- Q. What about adrenergic with epinephrine?
- RET
- Q. Give an algorithm for genetic testing in pheochromocytoma.
- Q. Should all the involved genes be tested at once?
- No
- Target gene approach to be applied as described above
- Q. Which genes to test in bilateral adrenal pheochromocytoma?
- Adrenergic- MEN 2 (RET)
- Noradrenergic – VHL
- Pearl
- Remember in RET- MTC is more common than pheochromocytoma – hence in the absence of MTC, MEN2 is less likely, however not impossible
- Q. Which gene to test first in case of abdominal paraganglioma?
- SDHB
- Then other SDH and VHL
- **Mode of inheritance **
- Q. What is the mode of inheritance of hereditary Paraganglioma syndromes?
- Most are autosomal dominant except SDHD and SDHAF2 which have the parent of origin effect- paternally inherited – paternal imprinting effect
- Other notes
- Q. What is the definition of malignant pheochromocytoma?
- WHO definition- the presence of distant metastasis
- In other tumors breach of the capsule defines malignancy
- Not so in pheochromocytoma
- Q. Which patients are more likely to have metastatic disease?
- extra-adrenal pheochromocytoma
- SDHB mutation
- Size >4-5 cm
- Q. What is the best way for drawing a plasma metanephrine sample?
- Supine for 20 min
- Patient resting before doing the test (no exercise)
- Q. What is the role of chromogranin A in Pheochromocytoma / PPGL?
- It is raised in 80% of cases
- However, it is not specific
- It is a useful tumor marker for follow-up of patients with metastatic disease
- PPI often increases CgA
- Q. Acetaminophen interferes with which assays?
- Liquid chromatography assays
- Stop for 1 week
- Q. Do SSRI /SNRI interfere ?
- Yes
- Q. Enlist the medication list which interferes with testing.
- Acetaminophen
- Sympathomimetics
- Tricyclic antidepressants
- Selective serotonin reuptake inhibitors
- Serotonin-norepinephrine reuptake inhibitors
- Levodopa
- Monoamine oxidase inhibitors
- Some beta-blockers (especially non-selective)
- Some alpha-blockers (i.e. phenoxybenzamine)
- Marijuana and other illicit drugs
- Q. Which is the best first-line imaging?
- Cross-section CT/ MRI is the best first-line test
- Q. Are all tumors MIBG avid?
- No
- Only 60% are MIBG avid
- 40% are not
- Q. What is the difference between Phenoxybenzamine vs prazosin?
- Phenoxybenzamine is non selective alpha blocker
- Prazosin is a selective alpha1 blocker
- Also phenoxybenzamine is irreversible binding while prazosin is reversible binding
- Q. What are the advantage and disadvantages of one over the other?
- Prazosin – can potentially be knocked off if there is a surge in catecholamine this can cause a problem in the case of an intraoperative period when a tumor is manipulated. This is not seen over phenoxybenzamine
- Phenoxybenzamine produces more problems post-op because since there is irreversible binding → new receptors need to be generated post-op to prevent hypotension. This is not seen with prazosin
- Q. What is the mechanism of action of tyrosine?
- It blocks tyrosine hydroxylase
- Q. Is metyrosine beneficial?
- Studies have shown less CV complication in metyrosine + Pheoxybenzamine vs PBZ alone
- Q. the regimen used in adults, is it the same as in children?
- Yes
- Q. When is cortical sparing surgery done?
- Experience surgeons who can do that for patients with bilateral adrenal disease or susceptibility to bilateral disease